Fibrosis quística como enfermedad en la adultez
Si bien la fibrosis quística (FQ) es una enfermedad genética predominantemente pediátrica, debe considerarse el aumento en la expectativa de vida y el diagnóstico en la adultez.
Estudio retrospectivo, descriptivo y comparativo
Objetivo: Analizar características genéticas y clínico-funcionales de adultos con FQ.
Métodos: Estudio retrospectivo, descriptivo y comparativo de 81 pacientes adultos con diagnóstico de FQ, asistidos entre 2013-2015; edad mediana: 26 años (rango: 18-61), divididos en Grupo A, diagnóstico de FQ antes de los 18 años y Grupo B, después de los 18 años. Se valoraron parámetros clínicos, genéticos, índice masa corporal (IMC), volumen espiratorio forzado en el primer segundo (VEF1), insuficiencia pancreática (IP), diabetes relacionada con FQ (DRFQ) y pancreatitis aguda (PA).
Resultados: Grupo A: 63 pacientes [edad mediana: 24 años (rango: 18-47)]. Mediana de edad al diagnóstico: 1 año. Media IMC 20,8 ± 2,1 DS m/kg2. IP 49/63 (77,7%). DRFQ 23 pacientes (36,5%), PA 2 pacientes (3,1%). Mediana VEF1 54 %. Estudio genético en 54/63, mutación más frecuente F508del (65/108 alelos).
Grupo B: 18 pacientes [edad mediana: 35 años (rango: 25-61)]. Mediana de edad al diagnóstico: 36 años. La media del IMC fue 23,5 ± 2,49 DE m/kg2. IP 6/18 (33%). DRFQ 1 paciente (5,5%), PA 1 paciente (5,5%). Mediana VEF1 66 %. Estudio genético en 15/18, mutación más frecuente F508del (14/30 alelos). Se halló diferencia estadísticamente significativa en la edad (p = 0,0001), el IMC (p = 0,0001), la IP (p = 0,0003) y el DRFQ (p = 0,01).
Conclusiones: Los pacientes con FQ diagnosticados en la adultez presentaron una edad media superior, mejor estado nutricional y un menor compromiso funcional pancreático.
Introducción
La fibrosis quística (FQ) es la enfermedad genética de herencia autosómica recesiva más frecuente en la población de origen caucásico, y está caracterizada por la disfunción de las glándulas de secreción exocrina debido a una mutación de un gen localizado en el cromosoma,(7) que codifica una proteína identificada como regulador de la conductancia transmembrana de fibrosis quística (CFTR).(1, 2)
La incidencia mundial es de aproximadamente uno cada 2500-3000 recién nacidos;(3) en nuestro país se estima en uno cada 6100 recién nacidos, según datos del programa de pesquisa neonatal. La prevalencia en portadores sanos de la mutación es de aproximadamente 1:40.(4)
La enfermedad respiratoria es responsable de la mayor proporción de morbilidad y mortalidad en FQ y constituye junto con la malabsorción la forma de presentación clínica más frecuente.(5)
El páncreas es el órgano del sistema digestivo más comprometido tanto en su función exocrina como endocrina: pancreatitis aguda (PA), pancreatitis crónica (PC) y diabetes relacionada con FQ (DRFQ).(5-7) Un 85-90% de los pacientes presentan insuficiencia pancreática exocrina (IP). La pancreatitis aguda constituye una complicación infrecuente de la FQ y se presenta principalmente en pacientes suficientes pancreáticos.7
Aproximadamente un 9,9% de los casos se diagnostica recién en la edad adulta.(8) En estos pacientes el compromiso pulmonar inicial es más leve y generalmente tienen un páncreas exócrino suficiente, a diferencia de aquellos en los que se diagnóstica FQ a temprana edad.
La sobrevida de los pacientes sigue en aumento, dando lugar a una población creciente de adultos que viven con FQ como una enfermedad crónica.(8) En Estados Unidos, en el año 2015, 51,6% de los pacientes eran adultos.(9)
El objetivo del presente trabajo fue comparar las características clínicas, genéticas, la función pancreática y la pulmonar de los pacientes con diagnóstico de FQ en la niñez con aquellos diagnosticados en la adultez, asistidos en la Unidad de Fibrosis Quística del Hospital Interzonal General de Agudos R. Rossi de la ciudad de La Plata (Argentina).
Material y métodos
Se realizó un análisis retrospectivo, descriptivo y comparativo de pacientes adultos = 18 años, asistidos en la unidad de FQ del Hospital Rodolfo Rossi de la ciudad de La Plata, desde enero de 2013 hasta marzo de 2015.
El diagnóstico de FQ se basó en criterios clínicos, test del sudor y/o detección de dos mutaciones del gen CFTR. Los pacientes fueron divididos para su estudio en dos grupos: grupo A: pacientes con diagnóstico de FQ en edad pediátrica (< 18 años) y grupo B: aquellos con diagnóstico de la enfermedad en la edad adulta (= 18 años).
Se registró: edad, sexo, edad al momento del diagnóstico, estudio genético, índice de masa corporal (IMC), volumen espiratorio forzado en el primer segundo (VEF1), amilasa sérica, elastasa fecal, glucemia en ayunas y a las 2 horas de una sobrecarga oral de 75 gramos de glucosa.
Se estableció la concentración de elastasa fecal < 200 µg/g para diagnóstico de insuficiencia pancreática (IP). El diagnóstico de DRFQ se estableció con glucemia en ayunas superior a 126 mg/dl y a las 2 h superior a 200 mg/dl.
El estudio genético incluyó la búsqueda de mutaciones mediante la metodología PCR ASO reversa; inno-lipa CFTR 19 y 17, innogenetics.
El diagnóstico de PA fue establecido por la presencia de dos de tres de los siguientes criterios:
1-dolor abdominal consistente con la enfermedad
2-amilasa sérica elevada más de tres veces del límite superior normal
3-imágenes fuertemente sugestivas [tomografía axial computarizada (TAC) contrastada, ultrasonografía (USG) o resonancia magnética (RMI)].(10)
Se confeccionó una planilla de recolección de datos con el software Microsoft Excel 2010. Los mismos fueron procesados mediante el programa EPI Info versión (7).
Los resultados fueron agrupados en tablas para facilitar su tratamiento y los valores se expresaron como media ± 1 DE o mediana con percentilos 25-75. Para la comparación de las variables cualitativas se utilizó el test de Chi2 o Fisher y T de Student para los cuantitativos. Se consideró diferencia significativa a una p < 0,05.
Resultados
Se incluyeron un total de 81 pacientes adultos (= 18 años) con diagnóstico de FQ, 48 varones (59,3%) (Figura 1), con una edad mediana de 26 años (rango: 18-61). De los 81 pacientes, 63 (77,8%) fueron diagnosticados en la edad pediátrica (Grupo A) y 18 pacientes (22,2%) en la edad adulta (Grupo B) (Figura 2).
Las características referidas a la afectación pancreática se exponen en la Tabla 1. La mutación detectada con mayor frecuencia fue F508del (deleción de fenilalanina en la posición 508A), que se presentó en el 57% de los alelos mutados. Las mutaciones y frecuencias detectadas en los 138 cromosomas estudiados en 69 pacientes se observan en la Tabla 2.
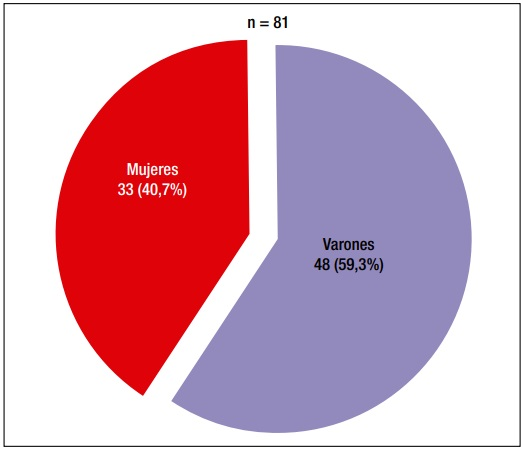
Figura 1. Distribución por género de la totalidad de los pacientes con FQ
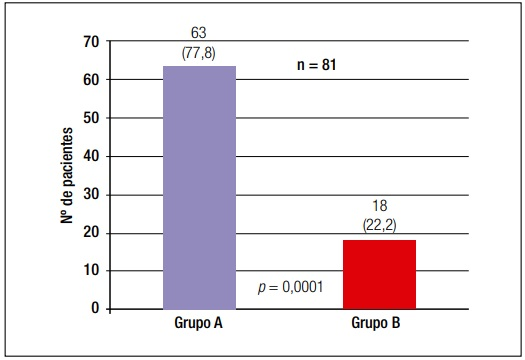
Figura 2. Distribución absoluta y relativa de los pacientes con FQ en ambos grupos
| Grupo A (n = 63) G | Grupo B (n = 18) | Total (n = 81) | |
| PA n (%) | 2 (3,1) | 1 (5,5) | 3 (3,7) |
| PC n (%) | 49 (77,7) | 6 (33,3) | 55 (68) |
| DRFQ n (%) | 23 (36,5) | 1 (5,5) | 24 (29,6) |
Tabla 1. Características de la función pancreática de los 81 pacientes con FQ.
| Mutaciones | n = 138 alelos (%) | Grupo A n = 108 alelos (%)* | Grupo B n = 30 alelos (%)* |
| F508del | 79 (57,2) | 65 (60,2) | 14 (46,7) |
| G542X | 10 (7,2) | 8 (7,4) | 2 (6,6) |
| R334W | 7 (5,1) | 3 (2,8) | 4 (13,3) |
| N1303K | 6 (4,4) | 4 (3,8) | 2 (6,6) |
| A455E | 2 (1,5) | – | 2 (6,6) |
| 2789 + 5G > A | 2 (1,5) | – | 1 (3,4) |
| R117H | 1 (0,7) | 1 (0,9) | – |
| R1162x | 1 (0,7) | 1 (0,9) | – |
| T3381 | 1 (0,7) | 1 (0,9) | – |
| 4016ins T | 1 (0,7) | 1 (0,9) | – |
| 3659delC | 1 (0,7) | 1 (0,9) | – |
| 3849 + 10kbc – T | 1 (0,7) | – | 1 (3,4) |
| 711 + 1G > T | 1 (0,7) | – | 1 (3,4) |
| Desconocidas | 25 (18,2) | 23 (21,3) | 3 (10) |
| CFTR: regulador de la conductancia transmembrana de fibrosis quística. *Valor absoluto en relación con el número total de cromosomas estudiados | |||
Tabla 2. Mutaciones del CFTR detectadas en los 138 alelos de los 69 pacientes adultos con FQ estudiados.
El grupo A presentó una edad mediana de 24 años (rango: 18-47). La edad mediana al diagnóstico fue de 1 año (rango: 0-15) y 28/63 pacientes (44,4%) eran mujeres. Presentaban IP 49 pacientes (77,7%). Desarrollaron PA 2 pacientes en la evolución de la enfermedad. Se manifestó DRFQ en 23 pacientes (36,5%). La media del IMC fue 20,8 ± 2,1 kg/m2. La mediana del VEF1 fue 54% (36-79). El estudio genético se realizó en 54/63 pacientes (85,7%), la mutación más frecuente fue F508del (65/108 alelos estudiados) (Tabla 3).
En el Grupo B la edad mediana fue de 35 años (rango: 25-61). El 27,7% (5/18 pacientes) eran mujeres. La edad mediana al diagnóstico fue de 36 años (rango: 20-61). Presentaban IP 6 pacientes (33,3%). En 1 paciente (5,5%) se diagnosticó PA en la evolución de la enfermedad. Se manifestó DRFQ en 1 paciente (5,5%). La media del IMC fue 23,5 ± 2,49 kg/m2.
La mediana del VEF1 fue 66% (35,5-107,75). El estudio genético se realizó en 15/18 pacientes (83,3%), al igual que en el Grupo A la mutación más frecuente fue F508del (14/30 alelos estudiados) (Tabla 3).
Al comparar las variables analizadas en ambos grupos se encontró diferencia significativa respecto de edad mediana (p = 0,0001), DRFQ (p = 0,01), IMC (p = 0,0001) e insuficiencia pancreática (p = 0,0003), mientras que el parámetro de función respiratoria VEF1, PA y la frecuencia del alelo F508del no mostraron diferencia significativa (Tabla 3).
Tabla 3. Comparación de las variables analizadas en ambos grupos en pacientes con FQ.
| Grupo A n = 63 | Grupo B n = 18 | p | |
| Edad años, mediana (rango) | 24 (18-47) | 5 (25-61) | 0,0001 |
| IMC (kg/m2 ) (x ± DE) | 20,8 ± 2,1 | 23,5 ± 2,4 | 0,0001 |
| VEF1 | |||
| (mediana y percentil 25-75%) | 54 (36-79) | 66 (35,5-107,75) | 0,2 |
| IP (elastasa < 200ug)n (%) | 49 (77,7%) | 6 (33,3%) | 0,0003 |
| F508del n | 65/108 alelos | 14/30 alelos | 0,1 |
| DRFQ n (%) | 23 (36,5%) | 1 (5,5%) | 0,01 |
| PA n (%) | 2 (3,1%) | 1 (5,5%) | 0,6 |
| Grupo A: diagnóstico en la infancia. Grupo B: diagnóstico en la edad adulta; DE: desvío estándar; IMC: índice de masa corporal; VEF1 : volumen espiratorio forzado en el primer segundo; DRFQ: diabetes relacionada con fibrosis quística. PA: pancreatitis aguda. | |||
Discusión
El estado nutricional se asocia fuertemente con la función pulmonar y la supervivencia en pacientes con FQ
En la actualidad la FQ ha dejado de ser una enfermedad exclusivamente pediátrica y con mayor frecuencia es mayor el número de enfermos que alcanzan la edad adulta, debido a que en los últimos años se ha incrementado la sobrevida de pacientes con la enfermedad.(11)
En el informe del recientemente creado Registro Nacional de Fibrosis Quística (RENAFQ) al 30 de mayo 2014, se incluyeron 692 pacientes con FQ confirmada por test del Sudor y/o estudio de mutaciones genéticas, de los cuales 156 eran mayores de 18 años (22,5%).(12)
Nuestro estudio muestra las características clínicas y genéticas de pacientes afectados con FQ cuyo diagnóstico se realizó en la edad adulta y los compara con pacientes también adultos, pero diagnosticados en la infancia. El porcentaje de pacientes diagnosticados de FQ en la edad adulta (Grupo B) representó el 22,2% con relación al número total de pacientes controlados en nuestra unidad.
Similar porcentaje fue documentado por De Gracia13 en España (20%). Si comparamos con otros estudios hallados en la bibliografía, nuestro grupo ha tenido una prevalencia menor en el diagnóstico de FQ en adultos que el obtenido por Fernández14 en Chile (38%) y sensiblemente superior al hallado por Lerin15 (14,8%).
El 68% de nuestros pacientes presentaba IP. Si bien clásicamente se reconoce que aproximadamente el 85-90%4 de los casos de FQ se asocian a IP, nuestro estudio pone de manifiesto que este elevado porcentaje podría estar relacionado exclusivamente con enfermos diagnosticados durante la infancia, ya que se estableció IP en el 77,7% de los pacientes del grupo A, y solo en el 33,3% de los pacientes diagnosticados de FQ en la edad adulta (grupo B), diferencia estadísticamente significativa (p = 0,0003) (Tabla 3).
El estado nutricional se asocia fuertemente con la función pulmonar y la supervivencia en pacientes con FQ.(16) En nuestra serie, hallamos que pacientes con FQ diagnosticados en la edad adulta (grupo B) presentaron mejores parámetros antropométricos que aquellos pertenecientes al grupo A (media de IMC 23,5 vs. 20,8m/kg2, respectivamente) (Tabla 3).
Los valores promedio de IMC hallados en los pacientes adultos del grupo B coinciden con los recomendados como objetivo nutricional a lograr para mantener una mejor función pulmonar de acuerdo con la Cystic Fibrosis Foundation.(17)
El gen CFTR abarca aproximadamente 180.000 pares de bases en el brazo largo del cromosoma (7). Actualmente se han identificado 2017 mutaciones.(18) La primera descripta, y la más frecuentemente hallada a nivel mundial es la mutación F508del, y se presenta a escala mundial en más del 70% de los alelos.3 En Argentina representa el 59% de las mutaciones documentadas.(19, 20)
El espectro de mutaciones en los latinoamericanos de origen europeo imita la de los principales países del sur de Europa, con una mayor frecuencia en los países que tienen un origen caucasoide. En nuestra serie, la mutación más frecuente fue la F508del (57,2% de los alelos mutados), al igual que los datos reportados en nuestro país.(19, 20)
La diabetes relacionada con la FQ (DRFQ) es la comorbilidad más frecuente.21 La nueva clasificación de la Asociación Americana de Diabetes (ADA) de 2014 incluye la DRFQ en el grupo de otros tipos específicos de diabetes secundarios a enfermedades del páncreas exocrino.22
La prevalencia aumenta con la edad, del 25% en sujetos mayores de 20 años y superior al 50% en pacientes mayores de 30 años.(23) La presencia de DRFQ se asocia con el deterioro acelerado de la función pulmonar, el estado nutricional y el aumento de la mortalidad.
La necesidad de una detección temprana de la alteración del metabolismo de la glucosa en la FQ es clara ya que está vinculada con el deterioro clínico.(24) La prevalencia de DRFQ en nuestra población (29,6%) fue inferior a la reportada por Adler A y col. (32,3%)(25) y Moran y col. (40-50%).(26) Se sabe que la DRFQ se presenta principalmente en pacientes con IP previa y parece ser más común en los homocigotos para la mutación del gen F508del,(4, 24) lo cual explica la mayor prevalencia en los pacientes del grupo A (36,5%) con respecto al grupo B, diferencia estadísticamente significativa. (p = 0,01) (Tabla 3).
La PA es una complicación infrecuente de la FQ. La prevalencia estimada de esta complicación en la población general de FQ es de 1,24%.(27) Tanto Durno(28) como Augarten(29) observaron esta complicación exclusivamente en suficientes pancreáticos.
En un estudio realizado en 2005 por De Boeck,(27) en 10.071 pacientes con FQ originarios de 29 países diferentes, la PA se presentó tanto en pacientes suficientes como insuficientes pancreáticos. En nuestra serie, la prevalencia de pancreatitis fue 3,7%, superior a la descripta en la literatura (1,76%-1,84%)(27, 28) y similar a la documentada por Sojo Aguirre (3,3%),(30) presentándose en forma exclusiva en pacientes con suficiencia pancreática.
En relación con la afección respiratoria, la disfunción del gen CFTR determina una disminución de la secreción de cloro e incremento en la reabsorción celular de sodio, produciendo secreciones viscosas y alteración de la depuración mucociliar. El deterioro progresivo es causado por la persistencia de la tríada inflamación, infección y obstrucción que conduce a la insuficiencia respiratoria y muerte.(31)
La prueba funcional respiratoria evidencia, al afectarse las vías aéreas centrales, la disminución progresiva del VEF1, clasificando de acuerdo con este parámetro el compromiso funcional respiratorio en leve > 70%, moderada 40-69% y grave < 39%.(4)
En nuestro estudio, aunque el grupo de pacientes diagnosticados con FQ en la edad adulta mostró valores discretamente superiores de VEF1, las diferencias no fueron estadísticamente significativas al compararlos con el grupo de diagnóstico en la niñez (p = 0,2) (Tabla 3).
En conclusión, en nuestra serie de pacientes en quienes se estableció el diagnóstico de FQ en la edad adulta se observó similar grado de compromiso funcional respiratorio, una mediana de edad superior, un mejor estado nutricional y un menor compromiso funcional pancreático, en comparación con pacientes en los cuales el diagnóstico de FQ se realizó en la infancia.
Autor: María Virginia D´Ascenzo, José Bosia, Claudio Girotti y colaboradores Acta Gastroenterol Latinoam 2018;48(1):06
Fuente: IntraMed
Referencias bibliográficas
1. Gregory RJ, Cheng SH, Rich DP, Marshall J, Paul S, Hehir K, Ostedgaard L, Klinger KW, Welsh MJ, Smith AE. Expression and characterization of the cystic fibrosis transmembrane conductance regulator. Nature 1990; 347: 382-386.
2. Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989; 245: 1066-1073.
3. Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med 2005; 352: 1992-2001.
4. Sociedad Argentina de Pediatría. Consenso Nacional de Fibrosis Quística. Arch Argent Pediatr 2008; 106: e1-e52.
5. Segal E. Consenso de fibrosis quística. Arch Argent Pediatr 1999; 97: 188-224.
6. Wilschanski M, Durie PR. Patterns of GI disease in Adulthood associated with Mutations in the CFTR gene. Gut 2007; 56: 1153-1163.
7. Kristidis P, Bozon D, Corey M, Markiewicz D, Rommens J, Tsui L, Durie P. Genetic Determination of Exocrine Pancreatic Function in Cystic Fibrosis. Am J Hum Genet 1992; 50: 1178-1184.
8. Nick J, Rodman D. Manifestations of cystic fibrosis diagnosed in adulthood. Curr Opin Pulm Med 2005: 11: 513-518.
9. Cystic Fibrosis Foundation Patient Registry 2015. Annual Data Report. [Internet]. 2015 [cited 2017 Jan 10]; Available from: https://www.cff.org/.
10. Banks P, Bollen T, Dervenis C, Gooszen H, Johnson C, Sarr M, Tsiotos G, Vege S, Classification of acute pancreatitis-2012: revision of the Atlanta classification and definitions by international consensus. Gut 2013; 62: 102-111.
11. Cabrera Roca G, Fernández-Burriel Tercero M, Cabrera Navarro P. Fibrosis Quística en la edad adulta: nuevas formas clínicas. Med Clin (Barc) 2003; 120: 584-588.
12. Grupo Registro Nacional de Fibrosis Quística – Argentina 2014. Fibrosis Quística: Informe del Registro Nacional. [Internet]. 2014 [citado Oct 22 2016]; Disponible en: http://www.sap2.org.ar/newsletter/enviados/informe%20fq.pdf.
13. De Gracia J, Álvarez A, Mata F, Guarner L, Vendrell M, Gadtner S, Cobos N. Cystic fibrosis in adults: study of 111 patients. Med Clin (Barc) 2002; 119: 605-609.
14. Fernández P, Labarca G. Fibrosis quística en el adulto: experiencia de un centro de referencia nacional. Rev Med Chile 2012; 140: 841-846.
15. Lerín M, Prados C, Martínez MT, Maíz L, Girón R, Solé A, Cabanillas J, Álvarez-Sala R. Fibrosis quística diagnosticada en edad adulta. Rev Clin Esp 2014; 214: 289-295.
16. Sheikh S, Zemel BS, Stallings VA, Rubenstein RC, Kelly A. Body composition and pulmonary function in cystic fibrosis. Front Pediatr 2014; 2: 1-7.
17. Stallings V, Stark L, Robinson K, Feranchak A, Quinton H. Clinical Practice Guidelines on Growth and Nutrition Subcommittee; Ad Hoc Working Group. Evidence-based practice recommendations for nutrition-related management of children and adults with cystic fibrosis and pancreatic insufficiency: results of a systematic review. J Am Diet Assoc 2008; 108: 832-839.
18. Cystic Fibrosis Mutation Database; editing status 2016-11-15; re3data.org – Registry of Research Data Repositories. http://doi.org/10.17616/R38356 last accessed: 2017-04-20.
19. WHO. The molecular genetics epidemiology of cystic fibrosis. Updated 2004. [Internet]. 2004 [cited 2015 Sept 28] Available from: http://www.who.int/iris/handle/10665/68702.
20. Oller de Ramírez AM, Ghio A, Melano de Botelli M, Dodelson de Kremer R. Fibrosis quística: diagnóstico molecular en 93 pacientes argentinos y detección familiar de portadores. Impacto asistencial y proyección a nuevos avances terapéuticos. Arch Argent Pediatr 2008; 106: 310-319.
21. Mackie AD, Thornton SJ, Edenborough FP. Cystic fibrosis-related diabetes. Diabet Med 2003; 20: 425-436.
22. Iglesias González R, Barutell Rubio L, Artola Menéndez S, Serrano Martin R. Resumen de las recomendaciones de la American Diabetes Association (ADA) 2014 para la práctica clínica en el manejo de la diabetes mellitus. Diabetes Práctica 2014; 5: 1-24.
23. Brennan A, Beynon J. Clinical updates in cystic fibrosis-related diabetes. Semin Respir Crit Care Med 2015; 36: 236-250.
24. Street ME, Spaggiari C, Ziveri MA, Rossi M, Volta C, Viani I, Grzincich GL, Sartori C, Zanzucchi M, Raia V, Terzi C, Pisi G, Zanetti E, Boguszewski MCS, Kamoi TO, Bernasconi S. Insulin production and resistance in cystic fibrosis: effect of age, disease activity, and genotype. J Endocrinol Invest 2012; 35: 246-253.
25. Adler AI, Gunn E, Haworth CS, Bilton D. Characteristics of adults with and without cystic fibrosis-related diabetes. Diabet Med 2007; 24: 1143-1148.
26. Moran A, Dunitz J, Nathan B, Saeed A, Holme B, Thomas W. Cystic fibrosis–related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care 2009; 32: 1626-1631.
27. De Boeck K, Weren M, Proesmans M, Kerem E. Pancreatitis among patients with cystic fibrosis: correlation with pancreatic status and genotype. Pediatrics 2005; 115: 463-469.
28. Durno C, Corey M, Zielenski J, Tullis E, Tsui LC, Durie P. Genotype and pancreatitis. Gastroenterology 2002; 123: 1857-1864.
29. Augarten A, Ben Tov A, Madgar I, Barak A, Akons H, Laufer J, Efrati O, Aviram M, Bentur L, Blau H, Paret G, Wilschanski M, Kerem BS, Yahav Y. The changing face of the exocrine pancreas in cystic fibrosis: the correlation between pancreatic status, pancreatitis and CF genotype. Eur J Gastroenterol Hepatol 2008; 20: 164-168.
30. Sojo Aguirre A, Martínez Ezquerra N, Bousoño García C, García Novo M, Heredia González S, Manzanares López-Manzanares J, Baranda García F, Vázquez Cordero C. An Pediatr (Barc) 2011; 75: 401-408.
31. Castaños C, Rentería F. Fisiopatología de la enfermedad respiratoria. En: Segal E, Fernández A, Rentería F. Fibrosis Quística. 1ª Ed. Buenos Aires. Ediciones Journal 2004: 79-100.
Realizar comentario
Noticias más leídas